Kenneth L. Scott, Gordon B. Mills, Timothy P. Heffernan and Lynda Chin are principal investigators participating in the Cancer Target Discovery and Development (CTD2) program administered by NCI’s Office of Cancer Genomics. Here, they describe their work to identify and characterize potential therapeutic targets from among the volumes of data being produced by large-scale genomics projects.
The Cancer Genome Atlas (TCGA), the Therapeutically Applicable Research to Generate Effective Treatments (TARGET), the Cancer Genome Characterization Initiative (CGCI), and the International Cancer Genomics Consortium, along with other molecular characterization projects, are cataloging genomic aberrations across major cancer lineages with the goal of identifying the most promising therapeutic targets and diagnostic biomarkers. The game-changing output from these projects promises to radically transform the way cancer science is conducted. At the same time, these efforts have revealed an extraordinary level of genome complexity made up of not only key “driver” events critical to pathogenesis, but also numerous biologically-neutral “passengers” that accompany unstable tumor genomes. The challenge now is to find ways to identify functional driver aberrations, as targeting such events or their activated pathways has the greatest hope of improving patient outcome.
This is a significant challenge, as the collective experience in target discovery and pharmaceutics has taught the community that computational analyses of genomics data alone are insufficient to identify new drug targets. Rather, successful drug development requires a thorough mechanistic understanding of a target’s cancer activity and the specific biological and genetic context in which it operates. Robust pipelines that allow prioritization of the thousands of potential targets are therefore critical for directing the research community’s limited resources toward the most promising driver candidates.
Unfortunately, the underdevelopment of functional genomics technologies has created a significant roadblock hindering our ability to rapidly assess the biological consequence of somatic aberrations in cancer. While RNAi-based screening platforms have been successfully used to validate new tumor suppressor genes, little progress has been made toward developing gain-of-function screening systems for validating over-expressed or hyper-activated oncogenes. These gain-of-function alterations are especially attractive given the proven efficacy of antibody and small molecule inhibitor therapies that target them.
To complement the power of RNAi-based approaches, our center within the Cancer Target Discovery and Development (CTD2) Network is implementing a scalable gain-of-function screening infrastructure aimed at accelerating functional validation of oncogenic driver events identified by TCGA and other high throughput sequencing efforts (Figure 1). Because each aberration within a given gene might result in a different functional impact or response to clinical therapeutics, we reasoned that it would be necessary to categorize every somatic event within each candidate gene through a combination of literature mining, predictive algorithms and functional characterization.
 Figure 1: The authors are principal investigators at one of the Centers of the Cancer Target Discovery and Development (CTD2) Network, whose mission is to bridge the gap between cancer genomics research and precision therapies to improve patient care
Figure 1: The authors are principal investigators at one of the Centers of the Cancer Target Discovery and Development (CTD2) Network, whose mission is to bridge the gap between cancer genomics research and precision therapies to improve patient care
To circumvent current technical bottlenecks limiting construction of numerous gene expression clones, Dr. Ken Scott’s research group (Baylor College of Medicine) devised a high-throughput mutagenesis and molecular barcoding (HiTMMoB) platform. The platform allows introduction of individual DNA mutations and small DNA insertions/deletions into a collection of over 32,000 sequence-verified human cDNA (or “gene”) clones, which were generated by the Orfeome Collaboration, Scott’s lab, and others. In addition to permitting efficient modeling of gene aberrations, HiTMMoB allows simultaneous “barcoding” of wild-type or mutant genes with unique 24-nuleotide fragments of DNA. Each barcode serves as a surrogate identifier for its associated gene, thus permitting detection and quantitation of driver genes in pooled gene screening strategies. Wild-type or mutant gene expression clones originating from HiTMMoB are subsequently entered into a series of screen-based assays designed to annotate their functionality as cancer drivers. Indeed, an important feature of this approach is that target cells, once made to express a wild-type or mutant gene, can be entered into parallel screens to maximize discovery potential (Figure 2).
Figure 2: A stepwise prioritization pipeline integrates computational and functional genomic approaches to identify alterations in cancer that are more likely to serve as therapeutic targets or biomarkers.
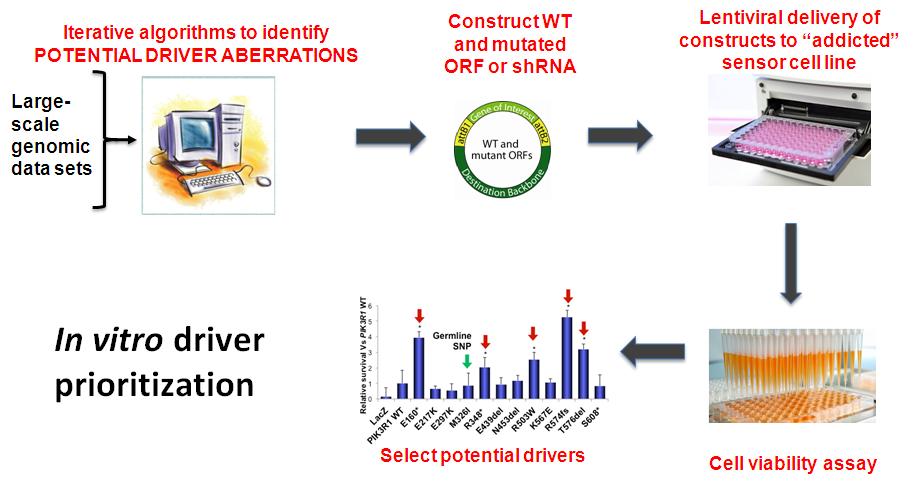
To do this, Dr. Gordon Mills’ laboratory (University of Texas M.D. Anderson Cancer Center) has implemented a sensor cell screening platform, based on the concept of “Transfer of Driver Addiction.” This platform quantifies the ability of the HiTMMoB expression clones to induce cell survival and proliferation, two key therapeutically targetable phenotypes. Specifically, the group employs the Ba/F3 myeloid cell system that is dependent on Interleukin-3 (IL3) for survival and proliferation. The goal of this approach is to assess whether addiction to IL3 can be transferred to a putative driver, an event detected by the ability of said driver to promote Ba/F3 proliferation in the absence of IL3.
An important feature of this strategy involves the ability to perform counter screens using a drug library containing “informer” therapeutic compounds targeting critical signaling pathways and cellular functions. IL3-independent growth drivers can then be assessed for their sensitivities to these agents to immediately provide clinically actionable information. This approach is linked to a high-throughput functional proteomics reverse phase protein array (RPPA) platform that can demonstrate functional consequences of each candidate gene. There is remarkable concordance between the therapeutic liabilities predicted by the informer drug screen and the functional consequences of the aberrant candidate gene as measured by RPPA.
This generalized Ba/F3 sensor system is used in parallel with other sensor cells, such as non-tumorigenic breast epithelial cells (MCF10A) and growth factor-dependent cancer cell lines, to identify drivers that alter viability and proliferation in different cellular contexts. Together, these in vitro approaches permit broad evaluation of driver candidates across multiple cancer lineages. Our initial studies demonstrate that the mutational context under which a candidate is evaluated has a marked impact on functional outcome.
While cell-based screening systems are tractable, in vitro models do not fully recapitulate all hallmarks of tumorigenesis and metastasis. To address this challenge, Dr. Lynda Chin’s team (University of Texas M.D. Anderson Cancer Center) developed a Context-Specific Screen (CSS) platform that interrogates the tumorigenic potency of candidate driver genes under the appropriate in vivo genetic and microenvironment contexts. These in vivo screens utilize genetically defined target cells (e.g., non-transformed human primary cells engineered with known signature genetic alterations) and are performed at orthotopic sites in the mouse (e.g., mammary fat pad sites for breast cancer gene candidates) to ensure the correct microenvironment context. Following orthotopic implantation of target cells virally-infected to express a pool of Dr. Scott’s barcoded candidate genes, genes driving tumor progression are identified from tissues by barcode amplification and sequencing. Enrichment for a driver event is defined as barcoded genes that are significantly higher in output (tumor or metastases) than input (injected cells), following the notion that tumors and metastases positively select driver genes and lose those with no role in tumor progression (i.e., passengers). Importantly, this pooled virus and barcoding approach permits discovery of cooperating driver events that are co-selected in output tumors.
In summary, our work will provide the greater research community multi-level functional assessments of oncogenomics data collected by TCGA and other large scale genomic studies. This level of technology development and biological annotation will create unique opportunities for transformative cancer research by directing the community’s efforts toward likely driver aberrations, thereby accelerating drug development and implementation.